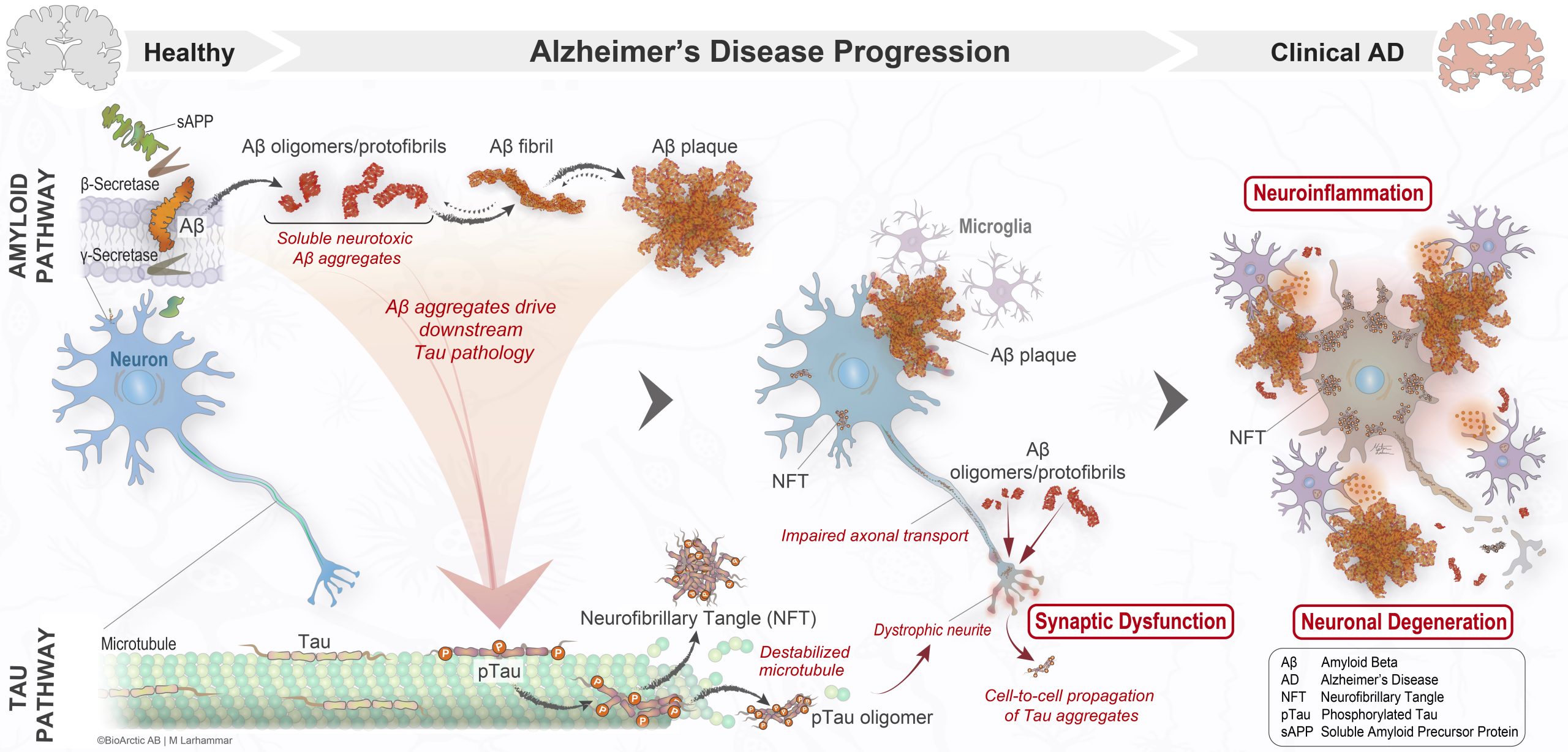
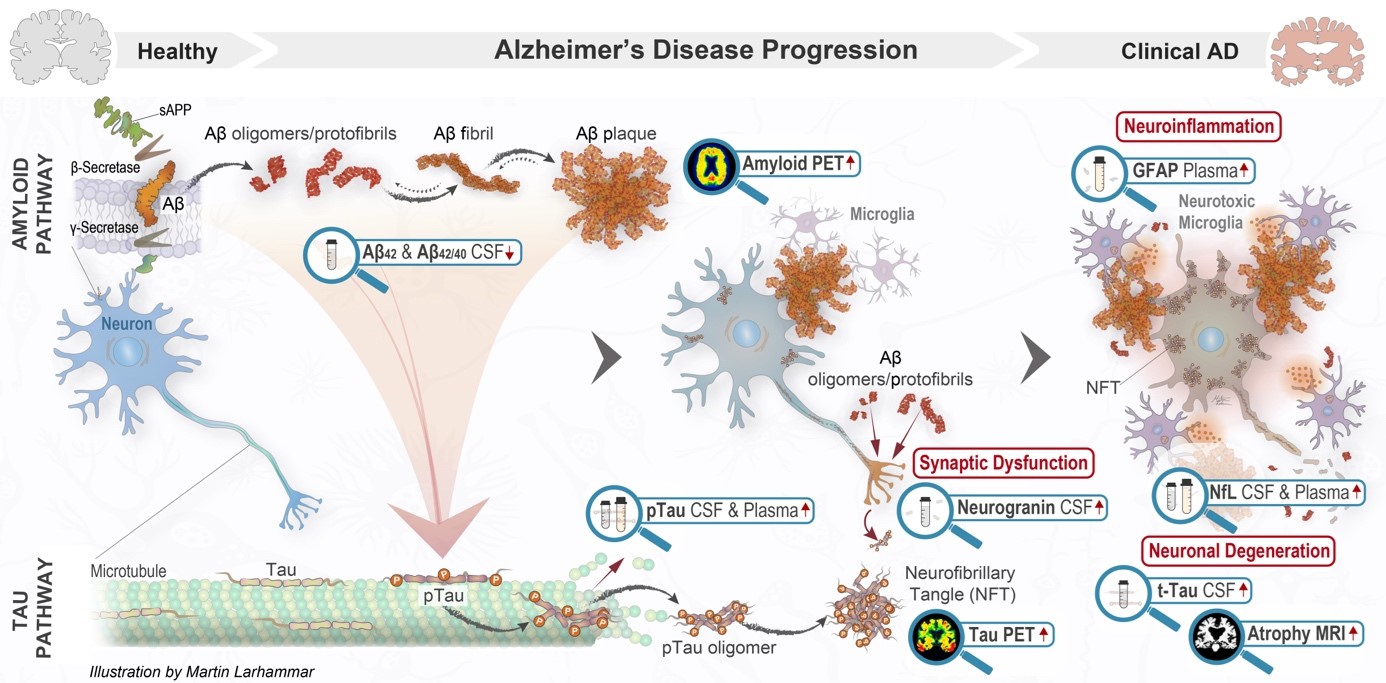
Referenser
1. Hampel H, et al. The amyloid-β pathway in Alzheimer’s disease. Mol Psychiatry. 2021 Oct;26(10):5481–5503. doi:10.1038/s41380-021-01249-0
2. Kumar A, et al. Alzheimer disease. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Aug 11 [citerad 2025 aug 20]. Tillgänglig från: https://www.ncbi.nlm.nih.gov/books/NBK499922/
3. Cummings J, et al. Alzheimer’s disease diagnostic criteria: practical applications. Alzheimers Res Ther. 2012 Sep 5;4(5):35. doi:10.1186/alzrt138
4. Jack CR, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018 May;14(4):535–62. doi:10.1016/j.jalz.2018.02.018
5. Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012 Aug 9;367(9):795–804. doi:10.1056/NEJMoa1202753
6. Hardy J, et al. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992 Apr 10;256(5054):184–5. doi:10.1126/science.1566067
7. Selkoe DJ, et al. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016 Jun;8(6):595–608. doi:10.15252/emmm.201606210
8. Hong W, et al. Diffusible, highly bioactive oligomers represent a critical species in Alzheimer’s disease pathogenesis. Acta Neuropathol. 2018 Jul;136(1):19–40. doi:10.1007/s00401-018-1846-3
9. Shankar GM, et al. Amyloid-β protein dimers isolated from Alzheimer’s brains impair synaptic structure and function. Nat Med. 2008 Aug;14(8):837–42. doi:10.1038/nm1782
10. Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002 Apr 4;416(6880):535–9. doi:10.1038/416535a
11. Hartley DM, et al. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999 Oct 15;19(20):8876–84. doi:10.1523/JNEUROSCI.19-20-08876.1999
12. Sehlin D, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012 Feb 15;7(2):e32014. doi:10.1371/journal.pone.0032014
13. Pooler AM, et al. Propagation of tau pathology in Alzheimer’s disease: identification of novel therapeutic targets. Alzheimers Res Ther. 2013 May 29;5(4):49. doi:10.1186/alzrt203
14. Tapia-Rojas C, et al. Mislocalized phosphorylated tau detaches from microtubules, destabilizes the cytoskeleton and impairs axonal transport. Alzheimers Dement. 2019 Feb;15(2):226–40. doi:10.1016/j.jalz.2018.08.006
15. Guillozet AL, et al. Neurofibrillary tangles, not senile plaques, parallel duration and severity of Alzheimer’s disease. Acta Neuropathol. 2003 Feb;105(2):148–53. doi:10.1007/s00401-002-0620-6
16. de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012 Feb 23;73(4):685–97. doi:10.1016/j.neuron.2011.11.033
17. Lewis J, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001 Aug 24;293(5534):1487–91. doi:10.1126/science.1058189
18. Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015 Apr;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
19. Streit WJ, et al. Dystrophic microglia in late-onset Alzheimer’s disease. Glia. 2020 Apr;68(4):845–54. doi:10.1002/glia.23782
20. McDade E, et al. Lecanemab in patients with early Alzheimer’s disease: detailed results of a randomized, double-blind, placebo-controlled, phase 3 trial. Alzheimers Res Ther. 2022;14:191. doi:10.1186/s13195-022-01033-9